
$48.5
Million
Truck Accident Case
We took more than 60 depositions in five different states. Because of Stephenson’s tireless advocacy, shortly before the trial was to commence, the defendants settled the case for $48.5 million.
Defective Medical Devices Lawyer
We’re fortunate in the United States that we have access to the miracles of modern medicine, and that includes medical devices that replace body parts or to otherwise treat or prevent disease. Many thousands are helped each year by medical devices – unfortunately, many thousands are injured when something goes wrong in the design, manufacture or implantation of defective medical devices.
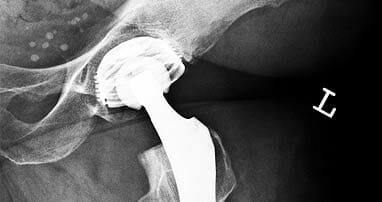
When that happens, the Indianapolis defective medical device lawyers at Stephenson Rife can step in to obtain fair compensation for those harmed by the devices, including bereaved families grieving the loss of a loved one killed by the failure of medical equipment. Call Mike Stephenson at 317-680-2501 for a free and confidential consultation.
What is a “medical device”?
A wide range of equipment used to diagnose and treat patients falls into the category of “medical device” – from simple tongue depressors to pacemakers and defibrillators. In addition, laboratory equipment, in vitro and test kits, ultrasound equipment, X-ray machines and medical lasers are classified as medical devices. Drugs and blood products are not considered to be medical devices.
Who regulates the manufacture and sale of medical devices?
Medical devices are among the products regulated in the U.S. by the Food and Drug Administration (FDA). Their Center for Devices and Radiological Health (CDRH) oversees firms who manufacture, repackage, relabel, and/or import medical devices sold in the United States.
Medical devices are classified into Class I, II, and III, with regulatory control increasing from Class I to Class III.
- Class I medical devices are simple in design and have little to no potential risk. Although they are not life-supporting or life-sustaining, they still must be registered and properly branded, labeled, manufactured and marketed. In Class I are items such as tongue depressors, elastic bandages, slings, splints, hand held instruments and examination gloves.
- Class II medical devices are more complex and may pose some risk to users. Most medical devices fall into this category, including hip and knee replacements, surgical mesh, X-ray machines, powered wheelchairs, infusion pumps, surgical needles and sutures, and acupuncture needles. Class II devices require special labeling, must meet mandatory performance standards and undergo post-market surveillance. They can be cleared for marketing through the streamlined 510(k) process.
- Class III medical devices are intricate in design and have the strictest guidelines because they pose the greatest risk. Before they are marketed, they must be scientifically reviewed in clinical trials and receive pre-market approval by the FDA. Class III devices include pacemakers, replacement heart valves, silicone breast implants, and implanted cerebral stimulators.
What is the 510(k) process?
To speed up the pre-market approval of Class II moderate-risk devices (and some Class III devices), Congress established an evaluation procedure commonly referred to as 510(k) approval. To gain FDA clearance through the 510(k) process, a medical device manufacturer must only demonstrate that their device is “substantially equivalent” to another device already approved for sale. They do not have to prove that the new device is effective, nor are they required to guarantee that it will not cause injury or death to patients.
The 510(k) process has come under scrutiny in recent years. In 2009, an Institute of Medicine committee was tasked by the FDA and Congress to review the 510(k) approval process. After a two-year study, they recommended that the process be replaced as it does not adequately protect the public from device failure, but the 510(k) procedure has not yet been abandoned.
What is a medical device failure?
Regrettably, medical devices don’t always perform as they were intended to. Sometimes this is due to a defect in the initial design of the product or its component materials; at other times, a manufacturing defect can cause the medical device to endanger the patient. And in other situations, the device may malfunction as a result of software failure.
When the FDA becomes aware that a medical device is defective or could be a risk to health, it may address the problem through a recall. You can read about some of the medical devices recently recalled by the FDA or withdrawn by the manufacturers.
Attorney Mike Stephenson
 Mike Stephenson has 40 years of experience and is a trusted advisor to many individuals and companies. His current practice is dominated by civil litigation in state and federal courts. He focuses much of his time on handling catastrophic injuries caused by all types of accidents, including motor vehicle, trucking, workplace injuries, product liability, and fire, just to name a few. He also works extensively in construction accidents. [ Attorney Bio ]
Mike Stephenson has 40 years of experience and is a trusted advisor to many individuals and companies. His current practice is dominated by civil litigation in state and federal courts. He focuses much of his time on handling catastrophic injuries caused by all types of accidents, including motor vehicle, trucking, workplace injuries, product liability, and fire, just to name a few. He also works extensively in construction accidents. [ Attorney Bio ]



